Lexikon der Chemie: Adsorption
Adsorption, ein Fall der Sorption, wobei die Anreicherung eines Stoffes an der Grenzfläche einer kondensierten Phase erfolgt. Den Prozeß Adsorptiv + Adsorbens
![]()
Adsorbat kann man analog wie eine chem. Reaktion betrachten. Die dabei freigesetzte Wärme wird Adsorptionsenergie (bei konstantem Volumen) oder Adsorptionsenthalpie (bei konstantem Druck) genannt. Man unterscheidet die reversible physikalische A. (Physisorption), die durch Desorption ohne chem. Veränderung von Adsorpt und Adsorbens wieder rückgängig gemacht werden kann, und die irreversible chem. A. (Chemisorption), obwohl eine scharfe Trennung nicht möglich ist. Bei der physikalischen A. wirken zwischen Adsorptiv und Adsorbens vorwiegend van-der-Waalssche Kräfte. Die molaren Adsorptionsenthalpien ΔAH liegen bei -5 bis -40 kJ mol-1 . Bei der chem. A. resultieren chem. Bindungen an der Oberfläche, und typische Werte für die Enthalpien liegen um -200 kJ mol-1. Reale Festkörperoberflächen sind energetisch und strukturell nicht homogen. Am Beginn der A. werden zunächst die energetisch günstigsten Oberflächenplätze von Adsorptivteilchen belegt, deshalb ist die Adsorptionsenthalpie vom Bedeckungsgrad abhängig. Für gleiche Bedeckungsgrade gemessene Adsorptionsenthalpien werden als isostere Adsorptionsenthalpien bezeichnet.
Im Idealfall setzt man voraus, daß die Oberflächen der Adsorbentien energetisch homogen und nach einer monomolekularen Bedeckung nicht mehr aufnahmefähig sind. Für den Bedeckungsgrad Θ gilt dann Θ = Γi/Γ∞, wobei Γi die Oberflächenkonzentration und Γ∞ die Sättigungskonzentration des Stoffes i darstellen. Γi ergibt sich als Quotient der adsorbierten Stoffmenge ni und der Oberfläche des Adsorbens Oi: Γi = ni/Oi.
Bei physikalischen A. stellt sich ein Gleichgewicht zwischen der Konzentration ci des Adsorptivs in der homogenen Phase und der Oberflächenkonzentration Γi des Adsorpts ein. Es ist temperatur- und druckabhängig. Die Abhängigkeit von Γi von der Konzentration ci oder dem Partialdruck pi (bei gasförmigen Adsorptiven) bei konstanter Temperatur wird als Adsorptionsisotherme bezeichnet Γi,T = f(ci bzw. pi), die Abhängigkeit des Γi von der Temperatur bei konstantem Druck pi als Adsorptionsisobare Γi,p = f(T). Die Adsorptionsisostere kennzeichnet die Konzentrationen ci, die bei verschiedenen Temperaturen zur gleichen Oberflächenkonzentration Γi führen: ci,T = f(T).
Praktisch am bedeutsamsten sind die Adsorptionsisothermen. Bei sehr geringen Bedeckungsgraden wächst die adsorbierte Menge zunächst fast linear mit der Konzentration, nähert sich aber für sehr hohe Konzentrationen einem Grenzwert Γ∞, der als monomolekulare Bedeckung aufgefaßt werden kann. Ein erneuter Anstieg kann durch die Ausbildung mehrerer Schichten des Adsorptivs oder durch eine Kapillarkondensation des Adsorptivs in Mikroporen des Adsorbens verursacht werden.
Für die mathematische Beschreibung der Adsorptionsisothermen von Gasen an Feststoffen existieren verschiedene Ansätze:
1) Freundlich-Adsorptionsisotherme Γi = αpβi (wobei pi Partialdruck des Adsorptivs, α, β Konstanten); 2) Langmuir-Adsorptionsisotherme Θ = αpi /(1+api); 3) Adsorptionsisotherme von Brunauer, Emmett und Teller (BET-Gleichung) Θ = αpi /(pio – pi)[1 + (α – 1) ·pi/pio]. Hierbei ist pio der Sättigungsdampfdruck des reinen flüssigen Adsorptivs. Diese Gleichung gilt bei Mehrschichtadsorption. 4) Die Gibbssche Adsorptionsgleichung gibt die an der Grenzfläche einer Flüssigkeit adsorbierte Menge eines Stoffes an:
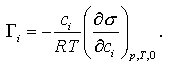
Hierbei ist ci die Konzentration des Stoffes i im Inneren der flüssigen Phase, R die allgemeine Gaskonstante, T die Temperatur in K, und (∂σ/∂ci) gibt die Änderung der Oberflächenspannung σ der Flüssigkeit bei der Veränderung der Konzentration des oberflächenaktiven Stoffes i an.
Bei der A. gasförmiger Verbindungen kondensieren bei Anwendung höherer Drücke und poröser Adsorbenzien die Gase und Dämpfe in den Poren (Kapillarkondensation).
Praktisch und technisch bedeutsam sind insbesondere Adsorptionsprozesse an hochdispersen, oft auch porösen Festkörpern mit großen spezifischen Oberflächen (z. B. Aktivkohlen, Molekularsiebe, Aluminiumoxid). Sie dienen z. B. zur selektiven Entfernung bestimmter Begleit- und Schadstoffe aus gasförmigen und flüssigen Mischungen (z. B. Entfärbung von Lösungen, Rückgewinnung von Lösungsmitteln, Entsalzung von Wasser, Rauchgasentschwefelung, Atemschutzfilter in Gasmasken), zur Beseitigung von Restgasen in Hochvakuumanlagen (Sorptions- oder Getterpumpen) und zur Stofftrennung (Parex-Verfahren). Auch in der heterogenen Katalyse spielt die A. der Reaktionspartner an der Katalysatoroberfläche eine entscheidende Rolle. Außerdem ist die A. ein Trennprinzip der Chromatographie. Die A. eines gelösten Adsorptivs an einem festen Adsorbenten spielt bei der Adsorptionschromatographie eine Rolle.







Schreiben Sie uns!